This webtool for primer selection is implemented as discussed in the paper about the Local Splicing Variation (LSV-Seq) Method:
Machine learning-optimized targeted detection of alternative splicing by Kevin Yang, Nathaniel Islas, San Jewell, Anupama Jha, Caleb M. Radens, Jeffrey A. Pleiss, Kristen W. Lynch, Yoseph Barash, Peter S. Choi (https://www.biorxiv.org/content/10.1101/2024.09.20.614162v1)
Given a list of ~50-100 base-pair RNA sequences to target for reverse transcription, the webtool will retrieve predictions for all possible primers, and predict the top DNA primers by predicted performance for each input RNA sequence. While “full” mode is more accurate, it focuses on targeting the > 190,000 pre-processed splicing events from the human GTEx dataset across 52 tissues. Splicing events are defined in terms of local splicing variations (LSVs), as defined in the original MAJIQ paper (https://elifesciences.org/articles/11752). “Lite” mode is more flexible, working for a variety of inputs that make it possible to target any transcriptome/any specified sequence of any species.
There are two primary use modes
Lite mode. More flexible and requires less tissue-specific information, though less accurate. Works for ANY transcriptome via FASTA file input. Also includes built-in ways to specify chromosomal coordinates for human and mouse transcriptome targeting.
Inputs allowed:
BED6 file specifying mouse or human chromosome coordinates, with columns 1 to 6 described here (https://genome.ucsc.edu/FAQ/FAQformat.html#format1). Note that the score in column 5 is not used and can be arbitrarily 1, for instance.
a FASTA file
a selection from a dropdown list of all target LSVs identified in the human GTEx dataset
Full mode. More accurate but requires more tissue- and experiment-specific information. Currently works for pre-processed human local splicing variations (LSVs) from the GTEx tissue dataset. LSVs are a way of conceptualizing splicing
Inputs allowed:
A selection from a dropdown list of all target LSVs identified in the human GTEx dataset
Both modes output a bed file in the same format:
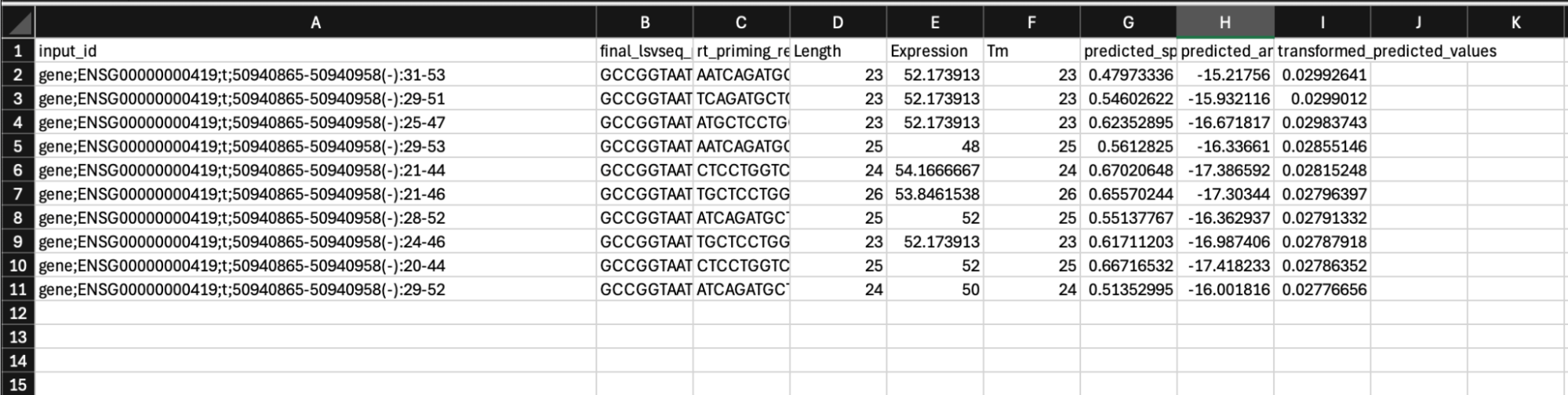
A list of the top 10 primer sequences for each region and their corresponding prediction scores, with information columns as follows:
“input_id” provides the name of the input region
“Final_lsvseq_primer” provides the LSV-Seq primer to order for convenience. The required additional primer sequence containing second sequencing adapter and in vitro transcription promoter is automatically added to rt_priming_reigon to provide the full LSV-Seq primer
“rt_priming_region” directly provides the reverse transcription primer sequence assessed by the model
“Length”, “Expression”, and “Tm" are primer length, target expression, and primer melting temperature respectively. “Expression” column is left empty for Lite model since it is not used in model predictions”
“Predicted_specificity_value”, s, is the specificity model prediction output. It should be interpreted as a proportion bound between 0 and 1. Higher is better.
“Predicted_amplification_value”, a, is the amplification model prediction output. It should be interpreted as a log value. Higher is better.
“Transformed_predicted_values” is the final combined value across the 2 model predictions used to rank the primers, specifically using the formula s*(λa). Lambda is set to 1.2 by default.
Evaluation of the performance of each mode is described in the paper.
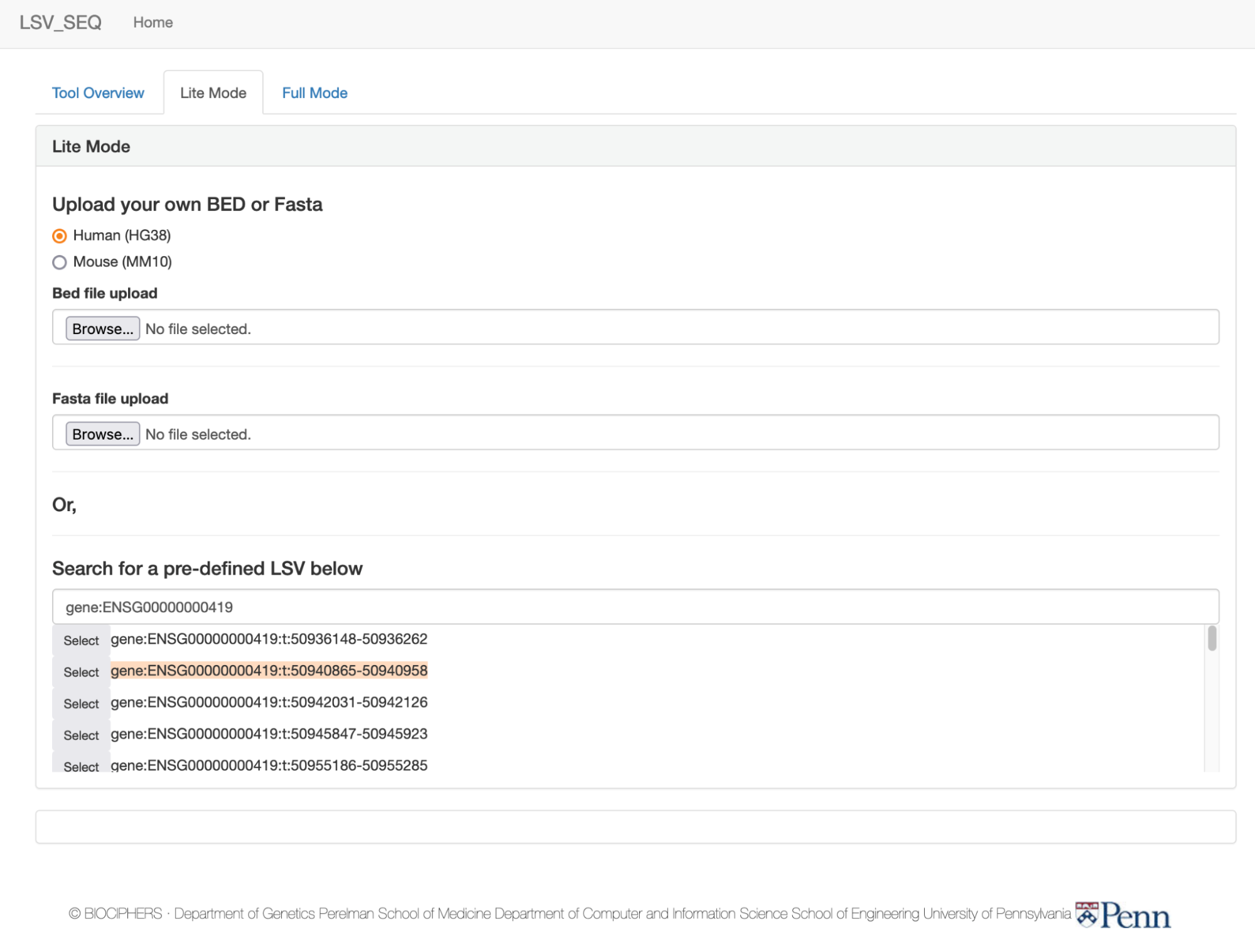
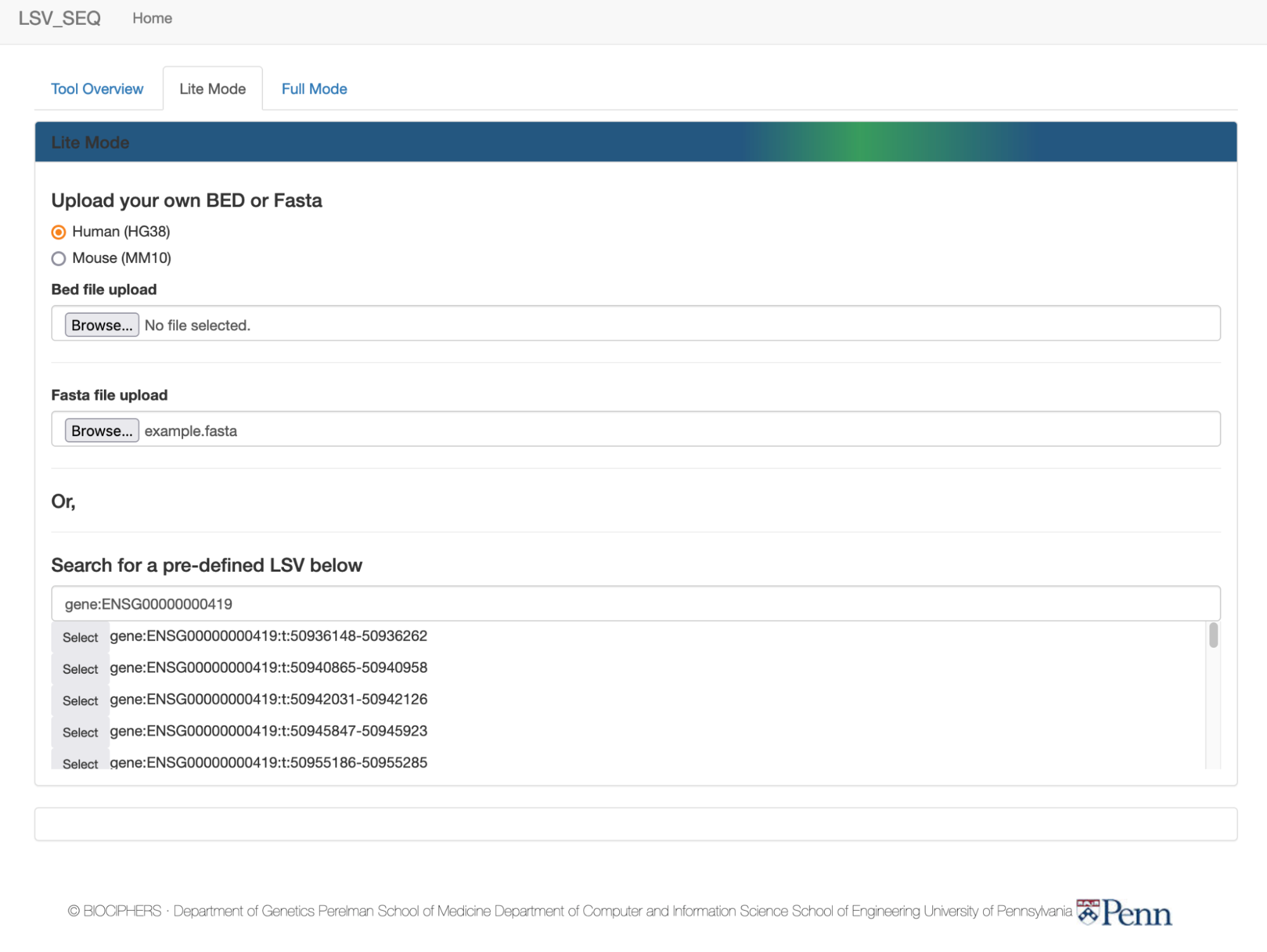
Upload your own BED or Fasta
Or,
Search for a pre-defined LSV below
First, choose or start typing a gene name or ID
Quickstart tutorial:
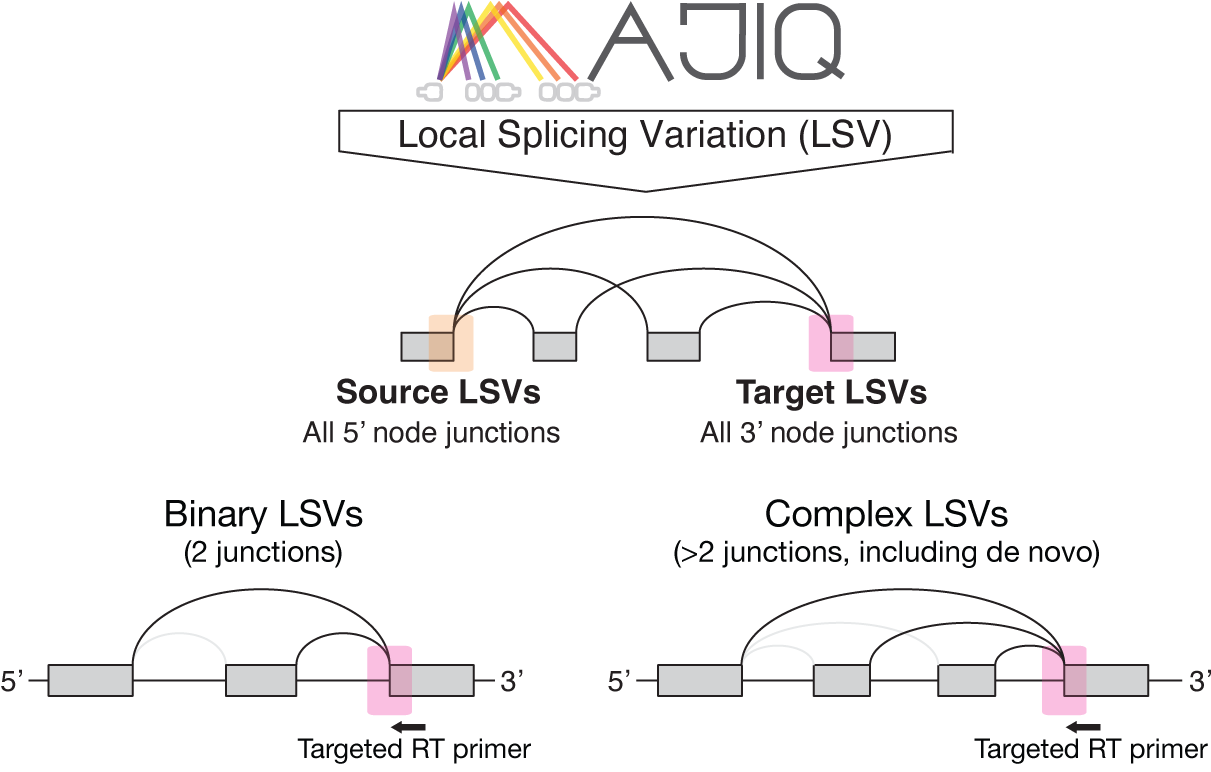
LSV nomenclature is formatted as “gene_id:t:reference_exon_coordinates”.
MAJIQ typically detects both single-target (:t:) and single-source (:s:) LSVs,
but LSV-Seq focuses on targeting single-target LSVs specifically (see
above diagram and
https://biociphers.bitbucket.io/majiq/lsv.html for more
details on the generalized LSV definition).
How to use the LSV dropdown menu:
Try typing ENSG00000000419 into the dropdown for labeled "enter gene name or id"
in the “Lite Mode” tab. It will
auto-populate to find the gene based on name or ensembl gene ID.
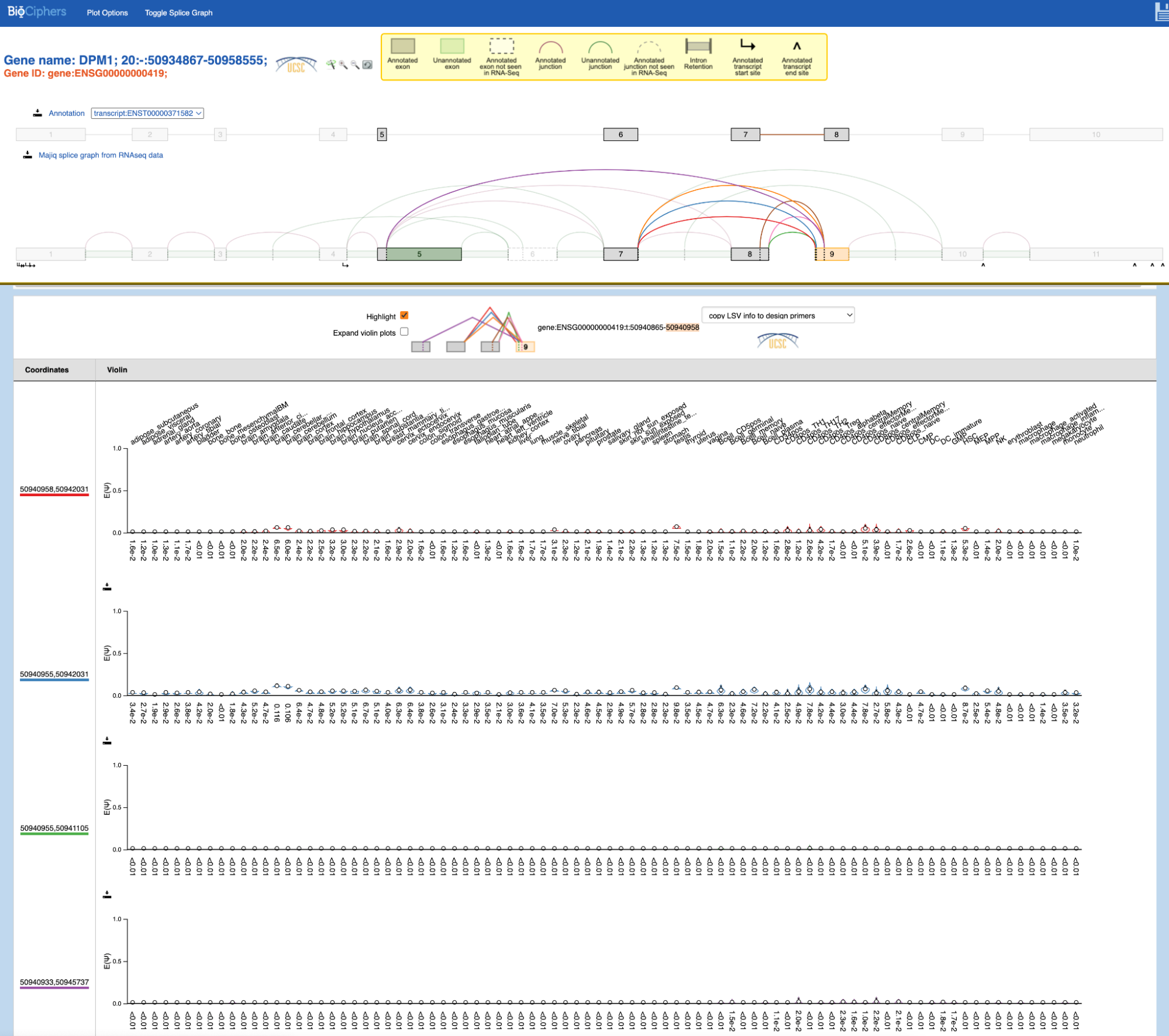
Click "select" next to the gene "DPM1", and the lower selector box will populate
with all known target LSVs sorted by reference exon
coordinates in the GTEx dataset. For
instance, the full LSV ID “gene:ENSG00000000419:t:50940865-50940958”
represents the single-target LSV in gene ENSG00000000419 with all
3’-node junctions entering the exon with coordinates 50940865-50940958:
Interpreting results:
Optimal Prime will automatically design a primer with maximal predicted score lying
within this 3’ exon and ensuring capture of all known 3’ splice site junctions. A TSV
will be automatically downloaded titled “final_output_primers_best.tsv”. Open the
resulting tab-delimited text into a word processor (i.e. Excel):
A list of the top 10 primers by region are ranked in descending order of final combined
score (as given by column “transformed_predicted_values”). The column “input_id” is the
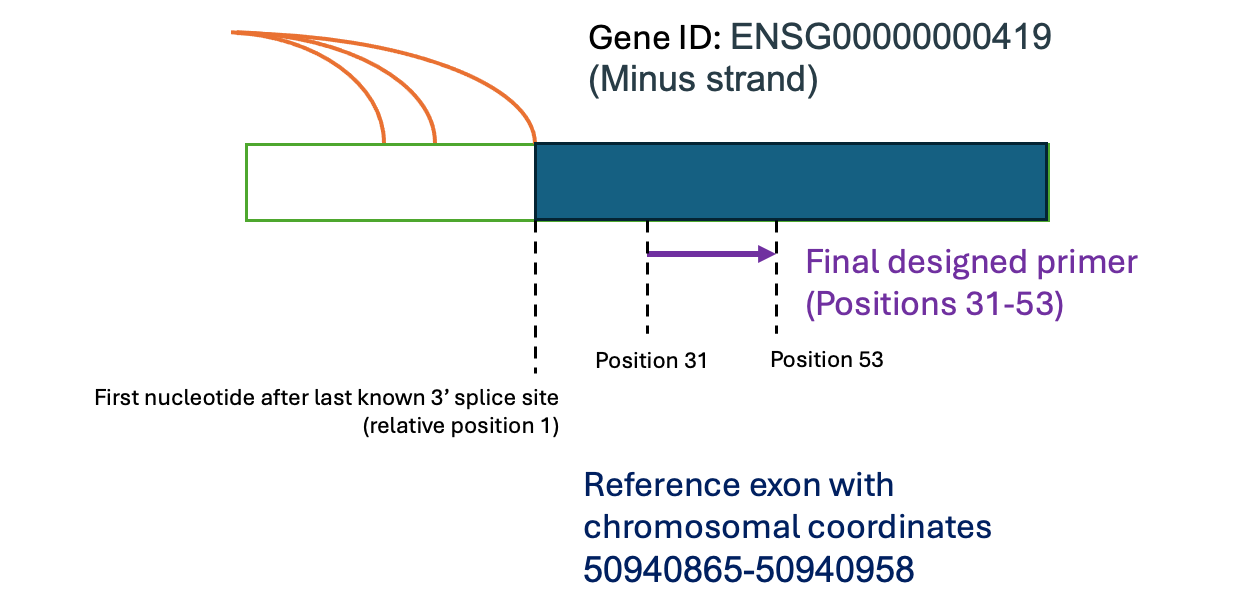
name of the primer, which is in format lsv_id(strand):start_position:end_position, where
“lsv_id” is the LSV ID, “strand” is + or - strand orientation of the gene,
“start_position”/”end_position” are the nucleotide positions relative to the 5’ exon
end. For instance, “gene;ENSG00000000419;t;50940865-50940958(-):31-53” represents a
primer designed for LSV ID gene;ENSG00000000419;t;50940865-50940958, which is on the
negative strand, with the output primer sequence corresponding to positions 31 to 53 of
the targeted 3’ exon, as schematized below.
The target-specific reverse-transcription primer sequence is given by the column
“rt_priming_region”, while the full-length LSV-Seq primer is given by the column
“final_lsvseq_primer“.
Please note that the tool will automatically truncate each considered region to the first 100 nucleotides
BED file upload:
Give a list of chromosomal coordinates in BED6 format corresponding to a list of 3’
exons to target (See
example.bed, which uses hg38. Columns in order: 1 = chromosome, 2 =
start coordinate, 3 = end coordinate, 4 = user-defined name, 5 = unused score column
(input 0), 6 = positive/negative strand). Be sure to select the correct genome (either
human hg38 or mouse mm10). The tool may take up to a few minutes to run before
downloading a final_output_primers_best.tsv.
FASTA file upload:
Give a list of AGCT nucleotide sequences in FASTA format corresponding to the targetable
region (See
example.fasta here). Can be any user-specified input name. No need to
specify chromosomal coordinates/strand or genome.
Code repository:
To access the code used to process the training dataset, extract the features, train the
Optimal Prime models, and run the final primer selection pipeline used in this web app,
please register at https://majiq.biociphers.org/optimalprime/app_download/.